
Estimated reading time: 14 minutes
The death rate from coronary artery disease (CAD) has declined considerably over the last three decades. This is mainly due to better control of risk factors and advances in therapy.
However, the prevalence of CAD remains high due to the aging of the population and better survival of those affected.
The current epidemic of obesity and type 2 diabetes will likely escalate the problem further.
Atherosclerosis, the underlying cause of CAD, is characterized by an accumulation of lipids, white blood cells, and cell debris in the inner layers of the arterial wall. The immune system is involved in the process, and inflammation appears to play a critical role (1).
Atherosclerosis can affect all arteries in the body but seems to have a high affinity for the coronary arteries.
Atherosclerotic lesions or plaques may protrude into the lumen of the coronary arteries, causing blockages that may limit blood flow to the heart muscle.
Additionally, rupture of an atherosclerotic plaque may cause thrombosis (blood clotting), completely blocking blood flow in a coronary artery. The result may be an acute heart attack.
The Role of Cholesterol and Lipoproteins
There is abundant evidence linking lipids, cholesterol in particular, with atherosclerosis (2).
In 1913, Nikolai N. Anitschkow, a Russian pathologist in Saint Petersburg, demonstrated that when given to rabbits, cholesterol, extracted from the egg yolks, (purified, and dissolved in vegetable oil) produced arterial lesions that closely resembled those of human atherosclerosis (3).
Many autopsy studies have shown a relationship between the amount of blood cholesterol and the extent of atherosclerosis (4). Furthermore, accumulation of cholesterol is found in human atherosclerotic plaques (5).
Because fats are insoluble in water, cholesterol cannot be transported in blood on its own. Instead, cholesterol is attached to hydrophilic proteins that function as transport vehicles carrying different types of fats such as cholesterol, triglycerides, and phospholipids.
These combinations of fats and protein are termed lipoproteins. Lipoprotein particles vary in the primary lipoprotein present and the relative contents of the different lipid components.
There is strong evidence that lipoproteins play a fundamental role in atherosclerosis and their interaction with the arterial wall appears to initiate the cascade of events that leads to atherosclerosis.
There are five major types of lipoproteins; chylomicrons, very-low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), low-density lipoprotein (LDL), and high-density lipoprotein (HDL).
Lately, the role of another lipoprotein, called lipoprotein(a) or Lp(a) has been highlighted (6).
Lipoproteins that promote atherosclerosis are termed atherogenic.
A specific protein, called Apolipoprotein B100 (ApoB), is an important component of all atherogenic lipoproteins (7).
Atherogenic lipoproteins such as LDL, VLDL, and Lp(a), all contain one ApoB molecule per particle. Hence, measurements of ApoB reflect the number of atherogenic particles.
On the other hand, HDL does not contain ApoB, and is not atherogenic. In fact, HDL appears to play a protective role and high levels of HDL particles are associated with less risk of coronary artery disease (9).
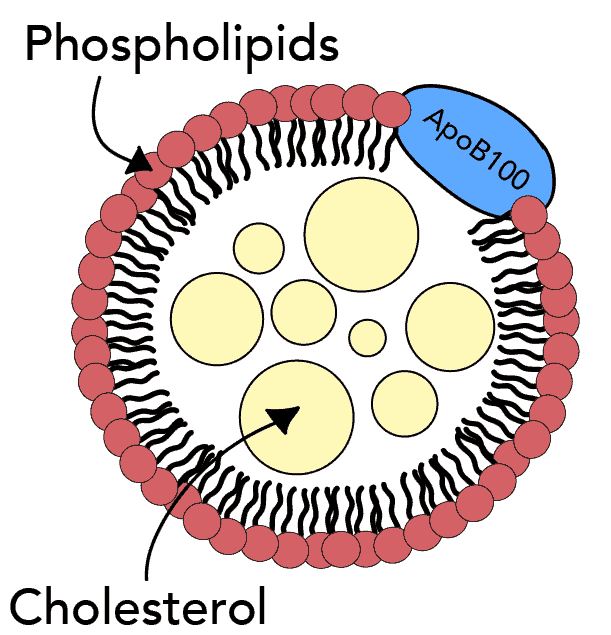
So, the atherogenicity of different lipoproteins is not determined by their cholesterol content. Instead, the lipoproteins’ molecular composition and the presence er absence of the ApoB molecule seem to be most important.
Lipoproteins are produced by the liver and their removal from the circulation is dependent on receptors found on the surface of cells, primarily in the liver. The LDL receptor plays a crucial role in the removal of LDL from the circulation.
Despite the proposed strengths of the associations between cholesterol, lipoproteins, and atherosclerosis, the underlying mechanisms have not been completely clarified.
Many people have high blood cholesterol throughout their lifetime without ever developing heart disease. Furthermore, a significant proportion of patients with coronary artery disease doesn’t have high blood cholesterol (10).
Retention of LDL in the Arterial Wall
The wall of an artery consists of three layers, the tunica intima, the tunica media, and the adventitia. Also, there is the endothelium, a thin cellular layer that lines the interior surface of the artery.
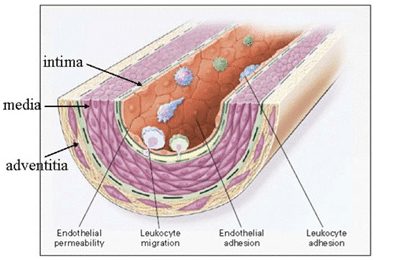 The intima and media consist of smooth muscle cells and extracellular matrix. The outermost layer, the adventitia, consists of looser connective tissue, nerve endings, mast cells, and vasa vasorum (a network of small blood vessels that supply the walls of larger arteries).
The intima and media consist of smooth muscle cells and extracellular matrix. The outermost layer, the adventitia, consists of looser connective tissue, nerve endings, mast cells, and vasa vasorum (a network of small blood vessels that supply the walls of larger arteries).
An accumulation of LDL in the arterial intima is an early step in atherosclerosis. Increased permeability of the endothelium and increased intimal retention of LDL are critical elements in the process.
LDL particles interact with particular constituents of the intima, notably the extracellular matrix. Here, the presence of ApoB may be a key factor.
Chondroitin sulphate proteoglycans (11) produced by smooth muscle cells in the arterial wall interact with ApoB on the surface of lipoprotein particles, thereby increasing the retention of LDL.
Endothelial dysfunction may also be a key factor. It may cause increased permeability of the endothelium allowing atherogenic lipoproteins to enter the vessel wall.
However, if lipoproteins are not retained in the intima, atherosclerosis is less likely to occur. Therefore, factors that promote lipoprotein retention are likely to induce atherosclerosis.
LDL Particle Size and Atherosclerosis
The size of the LDL particles may influence how readily LDL penetrates the endothelial barrier. Small LDL particles appear to penetrate the the endothelium and reach the intima 1.7-fold more than large LDL particles (12).
Also, small, dense LDL particles are more likely to bind to proteoglycans than large fluffy LDL particles (13).
High numbers of small, dense LDL particles are associated with increased risk for CAD in prospective epidemiologic studies. (14).
Small LDL particles are associated with high triglycerides (15), low HDL cholesterol (9), insulin resistance, metabolic syndrome (16), and type 2 diabetes.
The propensity of small LDL particles to be retained within the intima may explain why patients with metabolic syndrome and type 2 diabetes are at heightened risk of coronary artery disease in the face of normal or average blood levels of LDL cholesterol.
Modified LDL (Glycated LDL and OxLDL) and Atherosclerosis
Evidence suggests that to initiate atherosclerosis, LDL has to undergo chemical modification. Otherwise, it can not unlatch the typical cellular and inflammatory reactions typical of the disorder.
The oxidation hypothesis of atherosclerosis, summarized in 1989 by Steinberg, Whitman, and colleagues, suggests that oxidative modification of LDL plays a crucial role (17).
At the most basic level, oxidation is the loss of electrons. When a compound is oxidized, its properties change.
Because of its complex composition, the LDL particle is very sensitive to oxidized damage.
Each LDL particle contains approximately 700 molecules of phospholipids, 600 molecules of free cholesterol, 1600 molecules of cholesterol esters, 185 molecules of triglycerides, and one molecule of apoB. Both the protein and lipid moieties can undergo oxidative modification.
Whereas circulating LDL is reasonably stable, the long dwelling time of LDL within the intima provides a greater opportunity for oxidative modification.
Recent findings suggest that oxLDL begins to deposit in human coronary arteries before plaque formation and increasingly deposits with plaque growth (18).
Plasma concentration of oxLDL is associated with the risk of acute coronary heart disease events (19). One study found that plasma oxLDL was the strongest predictor of such events compared with a conventional lipoprotein profile and other traditional risk factors (20).
Oxidized LDL (OxLDL) promotes the immune and inflammatory reactions that characterize atherosclerosis. Evidence suggests that these reactions may be genetically determined (21).
Glycation is another type of atherogenic modification of LDL that may contribute to atherosclerosis (22).
Glycation is the result of bonding of a protein or lipid molecule with a sugar molecule, such as fructose or glucose, without an enzyme’s controlling action.
Small, dense LDL is more susceptible to glycation than more buoyant LDL (23).
Glycation and oxidation of LDL appear to be intimately linked and glycated LDL is more likely to be oxidized than non-glycated LDL (24).
Inflammation and Atherosclerosis
Inflammation plays a vital role in the formation of atherosclerotic lesions and the subsequent clinical complications (25).
Popular theories on the initiation of atherosclerosis suggest that modified lipoproteins, such as oxLDL, may play a central role in promoting the inflammatory reactions that characterize and drive atherosclerosis.
Cytokines are small proteins that are important in cell signaling (26).
Common cytokines include interferons, adipokines, interleukins, and tumor necrosis factor.
The discovery that vascular wall cells themselves can produce cytokines provided an important insight into the initiation of atherosclerosis.
According to the original concept, cytokines function to signal between leukocytes (white blood cells), hence the name “interleukin” (27).
Products of oxLDL may stimulate vascular wall cells to produce cytokines (28). These cytokines are believed to be mediators of inflammation and immune reactions in the atherosclerotic process.
Leukocytes, the type of white blood cells which is typically involved in most inflammatory reactions in the body, appear to play a significant role in atherosclerosis.
Leukocyte recruitment to the arterial wall is an important initial step in the formation of atherosclerotic lesions. The circulating leukocytes that enter the vessel wall are called monocytes, but within tissues, they are termed macrophages.
Typically, the endothelium resists the adhesion of leukocytes derived from blood. However, when stimulated by pro-inflammatory cytokines, adhesion molecules on the surface of endothelial cells may capture leukocytes (29). Hence, cytokines may play a key role in recruiting inflammatory cells in the vascular wall.
Failure of counter-regulatory mechanisms may also promote inflammation and oxidation in atherosclerosis. For example, HDL particles may function as carriers for anti-inflammatory and antioxidant mediators (30). In fact, HDL is an effective antioxidant.
HDL may also inhibit the expression of adhesion molecules in endothelial cells, thus reducing the recruitment of leucocytes into the artery wall.
Furthermore, HDL can inhibit the oxidative modification of LDL and thereby reduce the atherogenic potential of LDL.
Hence, low HDL levels may aggravate atherosclerosis because of blunted anti-inflammatory and antioxidant actions.
After reaching the intima, leukocytes (macrophages) take up modified lipoproteins. These-lipid laden white blood cells are called foam cells. Foam cells comprise the bulk of early atherosclerotic lesions, often termed fatty streaks (31).
Foam cells play a critical role in the occurrence and development of atherosclerosis.
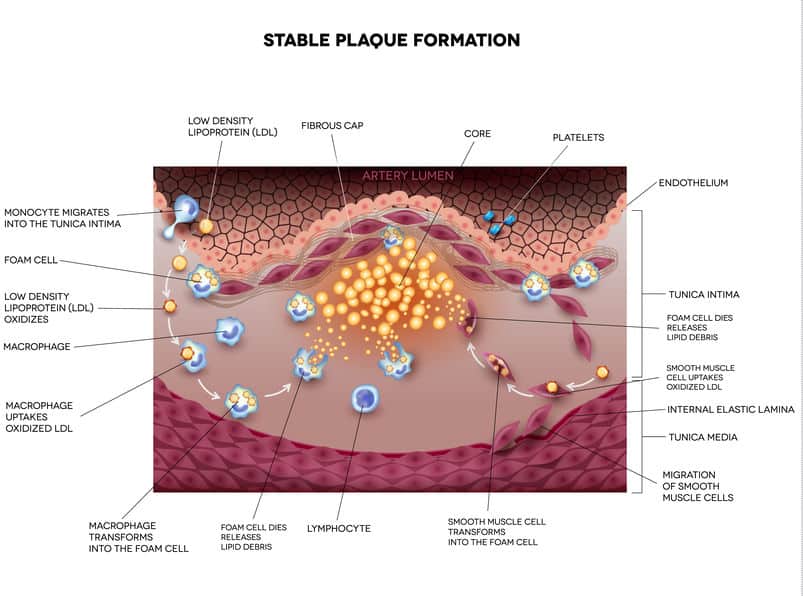
The Stable Plaque and the Vulnerable Plaque
Rupture of the plaque surface, often with superimposed blood clotting (thrombosis), frequently occurs during the evolution of coronary atherosclerotic lesions.
Actually, plaque rupture is an important mechanism underlying most cases of acute heart attack and sudden cardiac death (32).
The concept of plaque rupture was first reported at the autopsy of the celebrated neoclassical Danish artist Bertel Thorvaldsen, who died of sudden cardiac death in the Royal Theater in Copenhagen in 1844 (33).
However, it was not until the next century that researchers described the features of plaques responsible for acute coronary syndrome and sudden cardiac death.
Atherosclerotic plaques may become large over time and bulge into the lumen of the artery, limiting blood flow to tissues and organs. However, these plaques are not necessarily prone to rupture because the risk of plaque rupture depends on plaque type (composition) rather than plaque size (volume).
An atherosclerotic plaque that is prone to rupture is defined as a vulnerable plaque, whereas a plaque that is not prone to rupture is considered a stable plaque.
A vulnerable plaque is characterized by a thin fibrous cap, large lipid-rich necrotic core, plaque inflammation, increased vasa-vasorum vascularization, and intra-plaque bleeding (34).
One of the most significant challenges facing atherosclerotic research is identifying how and why plaques become vulnerable and how this may be translated into clinical practice.
The Role of Metabolic Syndrome and Insulin Resistance
Lately, metabolic syndrome, obesity, and type 2 diabetes have become increasingly common. These disorders are characterized by insulin resistance and an increased risk of CAD (16).
Insulin resistance is associated with low blood levels of HDL-cholesterol and high levels of atherogenic triglyceride-rich lipoproteins such as VLDL (35).
Increased availability of small LDL particles is common in people with insulin resistance (36).
Furthermore, insulin resistance is associated with higher levels of circulating oxLDL, which may contribute to atherosclerosis and acute coronary events (39).
The Limitations of Using LDL-Cholesterol to Assess Risk
The figure below is based on data from the Framingham Study showing the distribution of blood cholesterol in people with and without CAD (37). Both curves are bell-shaped with the top of the bell corresponding to the medium cholesterol level in each group.
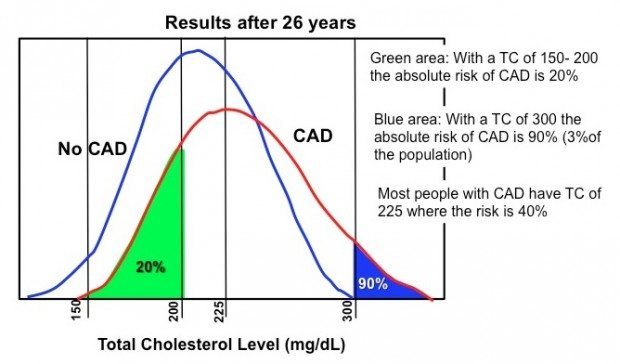
Notice that those who have coronary artery disease have slightly higher medium levels of blood cholesterol, but the difference is small.
Interestingly, a substantial number of people with normal cholesterol levels (<200 mg/dL) develop coronary artery disease. Furthermore, a significant number of individuals with elevated cholesterol (225-300 mg/dL) don’t have coronary artery disease.
Blood levels of LDL-cholesterol are commonly used to assess the risk of heart disease. However, using LDL-cholesterol to assess risk has several limitations (38).
The LDL-cholesterol value accounts for the total amount of cholesterol carried by LDL particles. Importantly, it doesn’t account for the number of LDL particles present in the circulation, which is more important when it comes to risk assessment (39).
Furthermore, LDL-cholesterol does not provide information about the size of LDL particles, which is important because small particles are more strongly associated with atherosclerosis than large particles.
So, of course, high blood cholesterol is not enough to cause atherosclerosis. But, if the availability of atherogenic lipoproteins such as LDL and VLDL is high, atherosclerosis is more likely to occur. However, for that to happen, other factors have to be present as well.
Lately, metabolic syndrome, obesity, and type 2 diabetes have become increasingly common. These disorders are characterized by insulin resistance and an increased risk of CAD.
A Few Practical Considerations
During the last 75 years, a simplified model of atherosclerosis and heart disease has been presented to health professionals and the lay public.
The model higlights cholesterol accumulation in the vessel wall as the main culprit. Consequently, lifestyle measures that lower cholesterol are emphasized.
Clinical and dietary guidelines have pinnacled LDL-cholesterol as an important target to prevent the occurrence of CAD.
Recommendations to limit the intake of saturated fats and cholesterol are based on the assumption that these types of fats will raise LDL-cholesterol and thereby increase risk. Hence, low-fat food products have been marketed with the aim of reducing the burden of cardiovascular disease.
Nonetheless, compared with low-fat diets, low-carbohydrate diets provide greater improvements in parameters associated with insulin resistance, such as HDL cholesterol, VLDL, LDL particle size, and particle number (40).
Moreover, low-carbohydrate diets provide greater reductions in inflammatory markers than low-fat diets (41).
There is no reason to believe that food products that elevate HDL cholesterol, lower triglycerides, reduce the availability of atherogenic LDL particles, and reduce insulin resistance and inflammatory markers, would be less effective in fighting heart disease than food that lowers LDL cholesterol.
That’s not to say that cholesterol doesn’t matter.
Lowering the availability of cholesterol-rich lipoproteins such as LDL and VLDL may be crucial in patients with coronary artery disease and individuals prone to atherosclerosis, such as those with familial hypercholesterolemia.
Denying the role of cholesterol in atherosclerosis is as naive as believing it explains everything.
Summary
An accumulation of LDL in the arterial wall is an essential step in the initiation of atherosclerosis.
Increased permeability of the endothelium and increased retention of LDL particles within the intima are important underlying mechanisms.
Small LDL particles are more likely to be retained in the intima than large buoyant LDL particles.
LDL particles may undergo chemical modification within the intima and become oxidized. OxLDL may enter white blood cells (leukocytes) called macrophages, which subsequently transform into foam cells. Foam cells are commonly found in atherosclerotic plaques.
Products of oxLDL may provoke vascular wall cells to produce cytokines, which promote recruitment of inflammatory cells into the vascular wall. Immune reactions and low-grade inflammation play a crucial role in the formation and progression of atherosclerotic plaques.
Rupture of the plaque surface, often with superimposed blood clotting (thrombosis), frequently occurs during the evolution of coronary atherosclerotic lesions. Plaque rupture is an important mechanism underlying most cases of acute heart attack and sudden cardiac death. Plaques that are prone to rupture are termed vulnerable plaques.
Currently, atherosclerosis is viewed as a complex multifactorial disorder involving the vessel wall, endothelial function, lipoproteins, lipoprotein modification such as glycation and oxidation, immune reactions, inflammation, and blood clotting (thrombosis).
It is the responsibility of experts in the field to educate health professionals and the general public about the complexity of atherosclerosis.
Unfortunately, the deep-rooted and oversimplified cholesterol-model of atherosclerosis has skewed recommendations on dietary interventions and other lifestyle measures to prevent coronary artery disease.
The article was initially published in 2016.
It was revised, updated and republished on February 13th, 2021.
Hi,
What do you think about Subbotin’s neovascularisation hypothesis of atherosclerosis? See https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3492120/
Subbotin is suggesting that the factors determining plaque formation are LDL-c and something else (which is why lowering LDL-c alone lowers the RR of CAD by an extremely variable amount).
RE Triglycerides: Studies by Dreon D et al showing associations between increasing carbohydrate intake & reducing particle size had 50% of the carbs as sugars. I declare shenanigans.
As Kitavans (~70%E from starchy carbohydrates) have high TG’s but virtually zero rates of CAD, it’s not TG’s that are the problem. It’s poor diet quality (high %E from over-refined foods) that’s the problem.
Cheers, Nige
Noige.
I believe Subbotin’s suggesting that lipoproteins don’t enter the intima through the endothelium but through new vessel formation (neovascularisation) from the vasa vasorum in the adventitia. Haven’t studies his theories in detail though.
Axel,
In https://jn.nutrition.org/content/134/11/3100.full , the upper tertile (~41ug/day) of Vitamin K2 intake had ~57% reduction in CHD mortality, compared to the lowest tertile (~15ug/day).
I don’t understand why a RCT on Vitamin K2 hasn’t been done, to test the hypothesis that Vitamin K2 drastically reduces atherosclerosis, by reversing calcification in the media. Do you know of any trials in the pipeline?
Suffice it to say, I supplement with Vitamin K2.
What form? MK-4? MK-7? Some say it makes a difference.
MK-4. It disappears from circulation into adipocytes much faster than MK-7. Other than that, I don’t think that there’s much difference.
It definitely works! See https://bit.ly/1PJFeel
All ldl’s begin their life as vldl’s don’t they ? And vldl’s are created in the liver and filled with trigs converted from carbs in the liver. Apparently in the process also polyunsaturated fats are packed in the vldl’s (Ralf Sundberg youtube video)
Reducing the amount of carbs converted in the liver should make the ldl’s fewer and bigger (less trigs, more cholesterol) ?
Reducing the amount of polyunsaturated fat should make the ldl’s less prone to oxidation ?
Why…..og why is saturated fat always convicted as the bad guy ?
You’re right Joern.
It is believed that most LDL is produced from VLDL.
Once in circulation, VLDL comes in contact with lipoprotein lipase in the capillary beds. Lipoprotein lipase catalyzes the breakdown of VLDL, releasing triglycerides for energy production or storage in adipose tissue. After the removal of triglycerides from VLDL, the composition of the lipoprotein changes and it becomes intermediate-density lipoprotein (IDL). Later, when the amount of cholesterol increases, IDL becomes low-density lipoprotein (LDL).
Thx for a brilliant summary by the way
I don’t think the “healthcare professionals” have figured out which came first… the inflammation or the cholesterol. I think when they figure out that the inflammation is first, and concentrate on what is causing the inflammation and that it could be a combination of different factors in different people they will solve the problem.
The big question might be: Do anybody want to «really» solve it, and more specifically: Who should fund the proper research.
Unless it will result in a drug there probably isn’t funding for it. And the statin business out of patent, but still a considerable industry will plummet, as will the new pcsk9 inhibitors industry not yet off the ground.
Almost 100 years of research and we have the current situation ?
Doc, I had a question while reading this analysis.
In my reading on VLC diets, there have been looks at North American Eskimos (pre-Western influence) and the Masai in Africa.
Most of the Masai all had atherosclerosis but the incidence of coronary infarct seemed almost nil.
Is it possible that atherosclerosis does not necessarily lead to heart attack? Perhaps it’s a normal part of aging?
Tanngl
Myocardial infarction (MI) usually doesn’t occur unless there is plaque rupture with superimposed thrombosis. So, you can have widespread atherosclerosis without MI occurring.
In many ways atherosclerosis can be regarded as a normal part of aging. We will all have it if we live long enough. So basically, it is premature atherosclerosis that we should regard as disease.
The lipoprotein transport system might not have been evolution’s greatest moment.
But evolution is primarily concerned with the survival of the species, not prolonged life expectancy
The LDL lipoprotein transport system prolongs life expectancy in the elderly, when infections are a major cause of death, whatever it might do in middle age.
I was referring to the mechanical construction of the lipoproteins. No doubt do they play an important and benificial role.
Evolution often deems things to be «good enough»
It can hardly foresee our deliberate attempt to destroy it….sugar, inflammation, Linoleic acid in excess etc
Thanks!
“So, of course, high blood cholesterol is not enough to cause atherosclerosis.”
I thought high LDL alone is sufficient. FH patients, anyone? That, of course, doesn’t mean that other factors aren’t involved or that a multifactorial approach (not just about lowering LDL) isn’t the best.
High LDL alone probably isn’t sufficient. Many individuals with very high LDL’s don’t develop CAD, including patients with FH. Of course, those with high LDL’s are at higher risk.
Axel, many individuals who smoke on a regular basis don’t get lunc cancer. Does that mean that smoking doesn’t cause lung cancer? 🙂
FH patients differ from others in one respect and yet their risk of getting CAD is much, much higher. So yes, high LDL alone is sufficient.
Ok Mie. Fair enough.. or maybe not.
You can’t really compare LDL with smoking. Smoking is an external/environmental factor that can hardly be compared to a biological substance, a lipoprotein that is necessary for normal physiological/biochemical equilibrium.
If you’re going to use the smoking analogy you would have to say that something that causes elevation of LDL causes atherosclerosis (for example some types of saturated fat).
I agree that there is abundant evidence and no doubt that smoking causes lung cancer. However, there is no clear evidence that external factors that raise LDL (such as some saturated fats) alone cause atherosclerosis (except maybe in animal models).
What I am trying to say (I suspect you’re deliberately missing the point) is that LDL will not lead to atherosclerosis unless other factors are present as well. For example, if we could reduce the inflammatory reactions associated with atherosclerosis, we could possibly reduce the atherogenicity of lipoproteins, thus rendering them less harmful. This hypothesis is currently being tested in the CANTOS trial; patients with CAD and elevated hsCRP (a marker of inflammation) are randomized to canakinumab (an anti-inflammatory drug that blocks the interaction of a certain cytokine with its receptor) or placebo.
“You can’t really compare LDL with smoking. Smoking is an external/environmental factor that can hardly be compared to a biological substance, a lipoprotein that is necessary for normal physiological/biochemical equilibrium.”
True. But, of course, irrelevant in this case. Of course, LDL per se is not the issue but abnormal levels of it. As for the elevation point, isn’t that a bit like splitting hairs? You might as well argue that it’s not smoking but something that CAUSES people to smoke, right?
“However, there is no clear evidence that external factors that raise LDL (such as some saturated fats) alone cause atherosclerosis (except maybe in animal models).”
Saturated fat may or may not raise LDL levels enough and it may or not come in packages (foods, that is) that don’t contribute to other risk factors. That is besides the point – which is that elevated LDL is a sufficient factor alone. Once again, FH patients.
“What I am trying to say (I suspect you’re deliberately missing the point) is that LDL will not lead to atherosclerosis unless other factors are present as well.”
I’m not missing the point. You are squirming to avoid my point. For instance, this
“For example, if we could reduce the inflammatory reactions associated with atherosclerosis, we could possibly reduce the atherogenicity of lipoproteins, thus rendering them less harmful (because less inflammation will lead to less atherosclerosis).”
is not any different from the current understanding. Dyslipidemia is of course an inflammatory condition too, and accumulation of LDL causes inflammation. So …? Reducing the reactions caused by this process would somehow indicate the LDL is not sufficient alone to cause CAD? Not anymore than e.g. getting people to wear seat belts (which reduce the impact of a car crash and thus health risks associated with it) would indicate that car crashes aren’t a health risk.
“Of course it is also theoretically possible that you can lower LDL to such an extent that it will not provoke atherosclerosis (because atherosclerosis will probably not occur in the absence of atherogenic lipoproteins). That’s another hypothesis that is currently being tested in the large clinical trials with PCSK9 inhibitors.”
There’s plenty of experimental evidence showing that LDL receptors work optimally when LDL levels are considerably lower than they currently are and that the risk is pretty much linear. And preliminary evidence from PCSK9 trials shows precisely that. Read e.g. this
https://wholehealthsource.blogspot.fi/2015/04/new-study-strengthens-case-that-ldl.html
The FH patients may be the equivalent of the cholesterol-fed rabbits. The rabbits were fed cholesterol well above their threshold of tolerance. FH patients may have LDL above a threshold of tolerance (total cholesterol levels of 650-1000 mg/dL are typical of homozygous FH), but that doesn’t prove that variations in the range below that threshold are meaningful.
The “may” there is misguided: the risk increases in a pretty much linear fashion among us “regular folk” too and is just as strongly a sufficient (and necessary) factor.
My understanding is that the linear association of LDL-C with CHD risk is weak and contingent in the normal range.
For example, in Hooper et al (2015), a meta-analysis of saturated fat reduced diet trials, lowering LDL-C by substituting PUFA (an essential nutrient(s) that is associated with vitamin E, another essential nutrient) for SFA (an inessential nutrient) was associated with a reduction in non-fatal CHD events (not including heart attacks). Whereas lowering LDL-C by substituting carbohydrate (an inessential nutrient) for SFA (an inessential nutrient) was not associated with any change in risk.
This is a weak association of LDL-C with risk in a real-life experiment, and it showed that what effect of LDL-C there was, was contingent on the means used to adjust LDL-C, not the LDL-C level itself.
“My understanding is that the linear association of LDL-C with CHD risk is weak and contingent in the normal range.”
Your understanding is … well, wrong. Or then you’re going for a semantic “argument” than a real one? “Normal” in western population is not really the same as “optimal”, you do know that?
“For example, in Hooper et al (2015) …”
The problem with this – and many other meta-analyses is that it doesn’t take into account the size of the effect. In RCT evidence, large trials like WHI which had statistically significant but clinically insignificant results tend to bias the view. Or then you’ve got underpowered studies that didn’t last long enough to show a benefit. Or both. If you, however, look at better quality studies like LA Veterans or Oslo Diet Heart where the changes were clinically significant, you get – surprise surprise – clinically significant results.
Concerning carbs: you do know that the abovementioned more or less applies to the question as well? And that LDL-C isn’t the only factor involved, which further attenuates the findings?
“This is a weak association of LDL-C with risk in a real-life experiment, and it showed that what effect of LDL-C there was, was contingent on the means used to adjust LDL-C, not the LDL-C level itself.”
Nope. There’s no evidence that the means are relevant (if you, of course, adjust for confounding factors), but it’s precisely the reductiong that counts.
By “normal” I meant “not homozygous FH” for a start. Normal is a genetic judgement in this context.
Secondly, when you read the Hooper paper, you will find that the associations I reported come from the subgroup analysis of cholesterol lowering, which was only associated with benefit (events but not MI or mortality) if due to PUFA substitution. This specifically excluded the WHI study.
In all the diet-heart studies, food quality and carbohydrate quality was improved in the intervention arms compared to the control arms. The reason is obvious – biscuits, cakes and other baked goods, and deep fried foods were a major source of saturated fat in the past, even more so than today, and you could not reduce saturated fat without reducing these foods and trying to replace them with nuts, fish, vegetables and so on.
Given these changes, all of these trials should have had clinically significant benefits. Why didn’t they?
In the LA veterans study, which you call “good quality”, vitamin E was 9.4-fold higher in the experimental group (22.6mg vs. 2.4mg) [5] and only 16.0% of the RDA in the control group, indicating a very inadequate intake and a substantial confounding variable in favour of the experimental group.
The Oslo diet heart study was even more confounded – I’ll quote from Steven Hamley’s analysis at https://www.stevenhamley.com.au/2013/12/the-oslo-diet-heart-study.html
The experimental diet was higher in omega 3. Meats and eggs were replaced with fish, shellfish and ‘whale beef’. The experimental group was supplied with “considerable quantities of Norwegian sardines canned in cod liver oil, which proved to be popular as a bread spread”. And also soybean oil has some plant omega 3
The experimental diet was higher in vitamin D, which is a more important source of vitamin D for Norwegians. The fish and cod liver oil supplied 610 IU of vitamin D
The experimental group was encouraged to eat more nuts, fruits, and vegetables; to limit animal fats; and to restrict their intake of refined grains and sugar
The experimental diet was lower in trans fats. Paraphrasing from Ramsden, et al: in the two decades before the ODHS, Oslo males had a 7-fold increased incidence of an initial heart attack, which coincided with a rapid rise in partially hydrogenated fish and vegetable oil margarines. In the control group, about 6.9% of total calories came from these margarines, whereas they were “entirely restricted” in the experimental group.
The means are definitely relevant. Hooper reports that trials with cholesterol lowering by CHO had no association with benefit, but if you think it’s the reduction, not the means, that matter, you have to explain away the means as “confounding variables”. Which is course they are. The associations with CVD of confounding variables – micronutrients, essential fats, activity, trans fats, smoking, education, weight, HDL-C, HbAic, TG:HDL-C, and so on – is greater than that of LDL.
That’s why it’s important to control for confounding variables – because they are more important than LDL-C.
“Secondly, when you read the Hooper paper, you will find that the associations I reported come from the subgroup analysis of cholesterol lowering, which was only associated with benefit (events but not MI or mortality) if due to PUFA substitution. This specifically excluded the WHI study.”
So
a) “only” events? 🙂
And b) the abovementioned points concerning the effect sizes, underpowered studies etc.?
If I don’t remember wrong, the absolute effect size (LDL lowering) in studies included by Hooper was 0,21 mmol/L. In comparison, in LA Veterans the reduction in the experimental group was around 20% of starting point of more than 5 mmol/L. Gee, wonder what might explain the differences … 🙂
“Given these changes, all of these trials should have had clinically significant benefits. Why didn’t they?”
How about you read what I wrote above?
“In the LA veterans study, which you call “good quality”, vitamin E was 9.4-fold higher in the experimental group (22.6mg vs. 2.4mg) [5] and only 16.0% of the RDA in the control group, indicating a very inadequate intake and a substantial confounding variable in favour of the experimental group.”
Now you just need to show that this would skewer the results. As you probably know, RCT evidence doesn’t support this interpretation. See e.g.
https://archinte.jamanetwork.com/article.aspx?articleid=217259
“The Oslo diet heart study was even more confounded”
But of course.
Nope. The points mentioned by the blogger are irrelevant in the sense that the key point here is REDUCTION in LDL levels, both statistically and clinically meaningful one, which leads to a benefit. That is the key, fat replacement is but one part of this.
“The means are definitely relevant.”
Why is there no evidence to indicate this, then? Can you point one single meta-analysis showing that means matter? Hooper et al. won’t do, due to reasons stated quite clearly earlier.
“That’s why it’s important to control for confounding variables – because they are more important than LDL-C.”
Err … as many of the variables results in excessive amounts of LDL-C, you’re mixing apples with oranges here. As for factors like HDL-C, I suggest you review the existing evidence: modifying HDL-C has been shown – at best – to be less important than lowering LDL-C.
HDL raising doesn’t work because the means are the wrong drugs.
If studies are underpowered, you’re looking for a weak effect. Plenty of effects (eg low carb for weight loss) are significant in small studies.
Modern epidemiology corrects for vit E specifically
Your vitamin E meta-analysis is a review of supplementation, not of induced deficiencies, of vitamin E, which seems to be what was produced in the LA veterans study.
To me this discussion sums up everything that is wrong with the lipid hypothesis. It may well be true for people with CAD, in the contingent fashion laid out brilliantly by Axel. This seems likely. But when people arguing for LDL as a measurement from a nutritional perspective try to argue away vitamin deficiency, this seems to me wholly typical of the way the LDL hypothesis has distorted nutritional advice, so that advising the entire population of the USA, and by extension the world, to avoid saturated fat as completely as possible becomes more of a priority for the DGAC, AHA etc than finding nutritious food to eat, as seen in the recent dietary guidelines for Americans.
With what results you see.
“so that advising the entire population of the USA, and by extension the world, to avoid saturated fat as completely as possible becomes more of a priority for the DGAC, AHA etc than finding nutritious food to eat”
I’d say this is not true. The DGAC recommends a variety of whole, fresh food mostly from plant. They don’t put that much emphasize on reducing SFAs per se.
And, why would anyone want to eat SFAs? (the long-chain one that is, the one from animal food). I’m not away of any evidence that they are beneficial to health. Quite the contrary. See :
Overfeeding polyunsaturated and saturated fat causes distinct effects on liver and visceral fat accumulation in humans.
“Both groups gained similar weight. SFAs, however, markedly increased liver fat compared with PUFAs and caused a twofold larger increase in VAT than PUFAs. Conversely, PUFAs caused a nearly threefold larger increase in lean tissue than SFAs. Increase in liver fat directly correlated with changes in plasma SFAs and inversely with PUFAs.”
Nutrient signaling: evolutionary origins of the immune-modulating effects of dietary fat.
“In a comprehensive literature review, we show that the body preferentially upregulates inflammation in response to saturated FA that promote harmful microbes. In contrast, the host opften reduces inflammation in response to the many unsaturated FA with antimicrobial properties.”
I’m not away of any evidence that SFAs are health-promoting. And quite a few that they could have a negative effect so the choice is obvious. If you want beneficial SFAs, eat fibers rich foods (plants) and enjoy the short chain one.
I’m perplex why you place so much emphasize on alternative hypothesis (inflammation, micronutrients deficiency, sugar) when these hypothesis are much less well supported than the lipid hypothesis. The lipid hypothesis is supported by virtually all lines of evidences (biological, ecological, experimental, drugs trials) and there is a strong convergence of the evidences supporting it. We can’t say so of any other alternative right now. I don’t understand why the lipid hypothesis skeptic want it so much to be wrong. No offense but that just looks to me like you guy just want to be able to say” ah ha! told you so” and pat your back for being more intelligent that everyone else if it ever comes to being proven wrong (which is not likely at this point).
The latest DGA specifically advises people undereating iron, choline, and vitamin D to get these nutrients from foods that are poor sources, such as wholegrains, and avoids mentioning the fatty animal foods which are the best sources of these nutrients. That is a nutritional failure, and it is solely due to a belief in the harms of saturated fat on the part of the DGAC.
Where are these harms in two large recent epidemiological studies, the EPIC-Netherlands study, or the Malmo Diet and Cancer study?
In the former, saturated fat is associated with less CVD, in the latter (using a 7-day food diary), there is no association with CVD (even the non-significant trends are in favour of SFA) and SFA is associated with less diabetes.
Why would you want to eat SFA? Because if you don’t, if you try to avoid it, you overlook some nutritious foods and are likely to replace them with rubbish. For example, the DGAC recommends a high-grain diet and states that half of this should be wholegrains. What about the other half?
Counsels of perfection and magical thinking are not substitutes for sound nutritional knowledge. The things we know about nutrition are more important than the hypotheses. It is not about the lipid hypothesis being wrong (I generally accept the version Axel gives), it is about the result of applying it at a population level – an epidemic of obesity and diabetes and unnecessary malnutrition and overfeeding, which promotes CVD more surely than butter ever could.
You have quoted from papers without supplying links, so I cannot do much about that.
“t is solely due to a belief in the harms of saturated fat on the part of the DGAC”
Not quite. The link between red meat and colorectal cancer still require caution on a population level. And they don’t say to not eat red meat but to simply reduce its consumption.
They don’t advice against eating poultry and fish either. I think the evidence clearly points in the direction of a plant-based diet with some animal product as being optimal. My own diet is between 70-80% plant and 20-30% animal product (mainly whole, unprocessed food) and I believe this to be in line with the best available evidence. I don’t need to supplement anything and don’t get much SFAs, dietary cholesterol and heme iron in my body which I believe are detrimental to health.
“Where are these harms in two large recent epidemiological studies, the EPIC-Netherlands study, or the Malmo Diet and Cancer study?”
Probably same short-coming as most studies i.e. homogenous population, baseline serum cholesterol already too high, too little difference in the amount of SFAs consumed between low and high quartiles, etc etc. I’m sure you agree that we should not put too much hope in epidemiological studies anyway.
“For example, the DGAC recommends a high-grain diet and states that half of this should be wholegrains. What about the other half?”
Not whole, as in white rice and white bread. Do you really expect north american to eat healthy all the time? Most of them don’t even get 2 servings of vegetable a day.
“it is about the result of applying it at a population level ” That would be an enormous challenge, regardless of what type of diet we would like to apply and goes beyond what a scientific organization can do.
“an epidemic of obesity and diabetes and unnecessary malnutrition and overfeeding”
There is much more than the DGAC behind this – why not blame the VERY obesegenic environment that we live in? The fact that it’s extremly easy to overeating junk food and get too much empty calories nowadays?
“You have quoted from papers without supplying links, so I cannot do much about that.”
I gave the title and I’m sure you know how google works… but here it goes :
Overfeeding polyunsaturated and saturated fat causes distinct effects on liver and visceral fat accumulation in humans.
Nutrient signaling: evolutionary origins of the immune-modulating effects of dietary fat.
(Apologies for not seeing your citation style)
“the link between red meat and colorectal cancer still require caution on a population level. And they don’t say to not eat red meat but to simply reduce its consumption.”
The US Dietary Guidelines tell pre-menopausal women are at risk of an iron deficiency and don’t eat enough red meat to eat anything “healthy” other than red meat; nothing they do list is a good source of iron. I shouldn’t have to explain why this is harmful nonsense.
They also consistently say to use low-fat dairy products and lean meat and avoid tropical oils. There’s no justification for this, but it is consistent with the version of the lipid hypothesis that focuses on LDL-C.
“I’m sure you agree that we should not put too much hope in epidemiological studies anyway.”
So much for the link between red meat and colorectal cancer.
“too little difference in the amount of SFAs consumed between low and high quartiles”
If a factor of two isn’t enough difference, what is?
“applying it [the lipid hypothesis] at a population level… would be an enormous challenge, regardless of what type of diet we would like to apply and goes beyond what a scientific organization can do.”
So how do you explain the drop in US SFA intake since 1970 so it is now almost exactly the 10% recommendation, or the increase in PUFA and carbohydrate? Was this caused by the obesigenic environment? Which in large part consists of people not eating the foods they used to eat because the media, the school authorities, the recipe books, the food manufacturers have all imposed restrictions on, specifically, the cholesterol and saturated fat content of these foods, since way back, and where they could they have added MUFA, PUFA and sugars, with what results we see.
Your references – , over-feeding muffins? This is relevant how?
The problem is that the high palmitate content of the palm oil is added to the palmitate generated by DNL from the carbs in this artificial hypercaloric state. Meanwhile, the PUFA is not stored to the same extent because its longer chain length means it is oxidised at a faster rate than palmitate.
However, if the SFA contained lauric acid or other medium chain SFAs, these would have been oxidised even faster than the PUFA.
https://ajcn.nutrition.org/content/72/4/905.full
So the choice of palm oil is selecting the (plant) SFA source most likely to store more fat than PUFA
However, fat stored from linoleic acid will also be less because a large part of the carbon from the PUFA (23%) will be used to make cholesterol in the liver; this will not happen to the palmitate. In the long term, this cholesterol might matter more than the neutral triglyceride fat in the liver.
“Carbon from linoleate and alpha-linolenate is recycled into palmitate and cholesterol in amounts frequently exceeding that used to make long chain polyunsaturates.”
https://www.ncbi.nlm.nih.gov/pubmed/14559071
Yet, because of the lipid hypothesis, the DGAC now wants Americans to consume even more liquid oil.
That’s their decision – the obesigenic environment supplies it, but the experts recommend more of it.
And of course the serum palmitate level will drop if fewer carbs are eaten.
This seems to be the fault in the Nutrient Signalling paper – carbohydrate is not mentioned as a source of serum FFA or the major controller of serum levels of palmitic and stearic acid that it is.
I don’t doubt that your own dietary preferences are appropriate for you, if you tolerate carbs and eat mostly whole food. Myself I don’t tolerate or like much grains, I do tolerate low-carb veges and fruits cooked or served with butter, coconut oil, olive oil or cream, and I don’t need to eat any more meat or consume any more dairy (nor, I’m sure, do I eat any more total fat) than I ever did on the old diet. All else being equal, there’s evidence that the fatty versions of dairy and red meat are no worse and may be healthier than the low fat versions.
Whole dairy foods are an important part of the traditional Mediterranean and adding them to the DASH diet didn’t seem to hurt recently.
It’s not about carnivory, the lipid hypothesis applies to coconut and palm oil and palm kernel oil and all sorts of dairy products. One can reduce one’s intake of carbohydrate while eating just as many plants as before or more. If it comes to that, a carnivorous diet has always been an excellent way to stay nourished while working out what you tolerate, for a few the answer will be meat, for others it’s a safe starting point.
Red meat’s not something I eat every day because I do care about my heme iron intake and I can’t give blood. I also prefer not to eat high-cholesterol food like brains or 3-egg omelette every day, but will certainly eat them without fear if they turn up as the best thing on the menu. I won’t use palm oil, but there’s lots of reasons for that (you only find it in the worst junk food here, it kills orangutans, and it’s high in palmitate without the more beneficial SFA found in dairy).
“The latest DGA specifically advises people undereating iron, choline, and vitamin D to get these nutrients from foods that are poor sources, such as wholegrains, and avoids mentioning the fatty animal foods which are the best sources of these nutrients. That is a nutritional failure, and it is solely due to a belief in the harms of saturated fat on the part of the DGAC.”
Please provide evidence that following the guidelines results in getting too little nutrients.
“Why would you want to eat SFA? Because if you don’t, if you try to avoid it, you overlook some nutritious foods and are likely to replace them with rubbish.”
I agree on the fact that you shouldn’t avoid certain foods solely because of their SFA content, e.g. yoghurt.
“So much for the link between red meat and colorectal cancer.”
It seems that you seem to praise or neglect e.g. studies on mechanism and cohort studies solely on the basis of your preset notions. “Cherry-picking” is the term here. The links is as solid in terms of scientific evidence as the link between smoking and lung cancer.
“So how do you explain the drop in US SFA intake since 1970 so it is now almost exactly the 10% recommendation, or the increase in PUFA and carbohydrate?”
Are you seriously trying to argue that the US population follows the guidelines? 🙂
“The problem is that the high palmitate content of the palm oil is added to the palmitate generated by DNL from the carbs in this artificial hypercaloric state. Meanwhile, the PUFA is not stored to the same extent because its longer chain length means it is oxidised at a faster rate than palmitate.”
I.e. if you eat too much SFA, there are problems. Or did you try to explain that away? If you did, you kinda … well, failed. 🙂
“However, if the SFA contained lauric acid or other medium chain SFAs, these would have been oxidised even faster than the PUFA.”
If you want to claim a benefit over PUFA, you need to show it. Trial evidence?
“In the long term, this cholesterol might matter more than the neutral triglyceride fat in the liver.”
Perhaps, or perhaps not. The link you provided offers no answer to this question. You’re grasping at whatever straws you think might possibly have a remote meaning of some sort.
“Yet, because of the lipid hypothesis, the DGAC now wants Americans to consume even more liquid oil.”
As partial replacement of less beneficial alternatives & in moderate quantities, yes. Care to explain the harms as shown in trial and/or epidemiological evidence?
“This seems to be the fault in the Nutrient Signalling paper – carbohydrate is not mentioned as a source of serum FFA or the major controller of serum levels of palmitic and stearic acid that it is.”
Can you not read? The purpose of the paper is to inspect FATTY ACIDS in this aspect.
“Whole dairy foods are a staple traditional Mediterranean food and adding them to the DASH diet didn’t seem to hurt recently.”
Indeed, if you’re talking about fermented dairy products (benefits include e.g. vitamin D,vitamin K, effects on gut microbiota which results in less cholesterol synthesis). Note that this is due to factors not related to SFA and to factors that attenuate the effects of SFA.
Nothing to counter the lipid hypothesis here – in fact, the issue of about diet-heart hypothesis which isn’t quite the same thing.
“HDL raising doesn’t work because the means are the wrong drugs.”
Could be, but YOU need to show it. It seems that the means are as right as any other possible means (diet, exercise) but when LDL is already low, raising HDL-C offers very little additional benefit.
“If studies are underpowered, you’re looking for a weak effect. Plenty of effects (eg low carb for weight loss) are significant in small studies.”
Apples and oranges. There are NO hard end-point low carb studies in CVD reduction, they’re all risk marker studies. Showing benefit in CVD events and deaths is very different from showing benefit in risk markers.
“Modern epidemiology corrects for vit E specifically”
Please indicate how.
“Your vitamin E meta-analysis is a review of supplementation, not of induced deficiencies, of vitamin E, which seems to be what was produced in the LA veterans study.”
George, please. If you want to study the benefits of vitamin E, you first look at supplementation studies where the factor in question is isolated.
“To me this discussion sums up everything that is wrong with the lipid hypothesis.”
I bet. To a denialist everything not supporting the argument that it’s everything but LDL (or, in your case, it seems that you’ve constructed a strawman of the lipid hypothesis).
“With what results you see.”
Do tell me what are the results. If you’re trying to say that Americans have followed dietary guidelines, you’re simply talking BS.
Let’s go back to the LA veterans study shall we?
That was the study where the control group was deficient in vitamin E.
Is there evidence that a deficiency of vitamin E is a factor in heart disease?
Inverse correlation between plasma vitamin E and mortality from ischemic heart disease in cross-cultural epidemiology.
“Essential antioxidants were determined in plasma of middle-aged men representing 16 European study populations, which differed sixfold in age-specific mortality from ischemic heart disease (IHD). In 12 populations with “common” plasma cholesterol (5.7-6.2 mmol/L) and blood pressure, both classical risk factors lacked significant correlations to IHD mortality, whereas absolute levels of vitamin E (alpha-tocopherol) showed a strong inverse correlation (r2 = 0.63, P = 0.002). Evaluating all populations, cholesterol and diastolic blood pressure were moderately associated, but their correlation was inferior to that of vitamin E. In stepwise regression and multiple regression analysis, mortality was predictable to 62% by lipid-standardized vitamin E, to 79% by vitamin E and cholesterol, to 83% after inclusion of lipid-standardized vitamin A (retinol), and to 87% by all the above parameters plus blood pressure. Thus, in the present study the cross-cultural differences of IDH mortality are primarily attributable to plasma status of vitamin E, which might have protective functions.”
In the LA veterans study, the “high quality” trial you mentioned earlier in this discussion, the intervention group had a significantly lower rate of CHD mortality. So far, so good, whatever the reason. But there was no benefit in terms of overall mortality from preventing these heart attack deaths, because the rate of cancer and other non-cardiac mortality increased in the intervention group. CHD isn’t the only game in town, there are clearly other ways to die, and more of us may find out what these are if we heed the new DGAC advice to double down on oils.
Have Americans followed dietary guidelines? Their SFA intake has declined to within a % of that recommended. I’d say mission achieved.
The Guidelines didn’t originally have a limit on sugar – they were all about replacing fat with carbohydrate. This was done.
Americans, on average, only fail to follow the guidelines in so far as the guidelines keep changing. But Americans (especially white Americans) are actually quite good, on average, at following the guidelines that were promulgated at some earlier date. The NHS data clearly shows this trend. See figure 1
https://aje.oxfordjournals.org/content/161/7/672.full
“Let’s go back to the LA veterans study shall we?”
Let’s not, as you simply cannot admit to being wrong. Simple: if vitamin E was the factor confounding the results, you’d expect that there’d be consistent and solid evidence from trials focusing SOLELY on vitamin E. There isn’t.
“Have Americans followed dietary guidelines? Their SFA intake has declined to within a % of that recommended. I’d say mission achieved.”
If reducing SFA intake was a) the ONLY factor and if b) it was achieved by replacing SFA with vegetable oils, nuts, fruits, veggies and whole grains while c) not increasing energy intake.
This hasn’t happened.
Yes, let’s not go back. Your vitamin E argument may be fallacious, but it probably has little relation to the failure of LA veterans to reduce mortality due to an increase in non-CHD mortality in those fed PUFA.
The dietary guidelines have influenced the diets of Americans. This is not deniable. But as you haven’t read the various guidelines, you wouldn’t know that they haven’t recommended “healthy diet patterns” until very recently. For decades the thrust was on replacing SFA with CHO and PUFA. This was definitely done. People ate more as a consequence. The guidelines are a moving target, and true believers insist that they have always believed the same thing, but the historical record shows that the guidelines have in fact been changed with each fresh failure.
They are now so confused as to be impossible to follow, making it even easier to blame the public for not following them.
“Your vitamin E argument may be fallacious, but it probably has little relation to the failure of LA veterans to reduce mortality due to an increase in non-CHD mortality in those fed PUFA.”
MY argument? Err George, FYI: you made the argument that vitamin E was a key player here. I pointed out that there’s no evidence to back that up. As far as tot. mortality is concerned, it’s useless in trials of this scope, field and magnitude – far too crude a tool.
“The dietary guidelines have influenced the diets of Americans. This is not deniable.”
Semantic play on the word “influence”? So far Americans have FAILED to observe to key messages of guidelines, decade after decade.
“But as you haven’t read the various guidelines, you wouldn’t know that they haven’t recommended “healthy diet patterns” until very recently.”
Entire patterns, yes, you’re correct.
“For decades the thrust was on replacing SFA with CHO and PUFA. This was definitely done.”
The focus was NEVER to replace SFA with Western carbs.
“People ate more as a consequence.”
Nope, regardless of that. They didn’t include more fruits, veggies and berries in any meaningful amounts to replace SFA and bad carbs.
“The guidelines are a moving target, and true believers insist that they have always believed the same thing …”
Err George? More argumentation, less storytelling, please.
“They are now so confused as to be impossible to follow, making it even easier to blame the public for not following them.”
Impossible? I agree. But only because people prefer fast food and other junk over healthy alternatives.
“The focus was NEVER to replace SFA with Western carbs.”
I must have missed the bit where it said to only replace SFA with rice.
If they didn’t want people to replace SFA with sugar and refined grains, they should have listed those as “nutrients of concern” as well and recommended limits. That’s the way guidelines work. In fact they assumed that any substitution of CHO for SFA, because it would lower LDL, would improve the health of the population.
That was the thinking; that was the important point of the original DGA; to reduce heart disease by lowering LDL across the population.
That’s why we have low-fat food with sugar in it – to help people to meet that goal.
my aunt smoked all her life. died of something else at age 80. my friend smoked, died at 60.
George, I will reply here because I can’t directly reply under your post in this thread.
I’d like to ask a different question :
Why do you think we should emphasize saturated fat?
I’m not aware of a any evidences showing SFAs (the long chained one, from animal product) to be beneficial to health. If anything, they are neutral, but there is other lines of evidences which clearly makes them seem bad health wise. See
Overfeeding polyunsaturated and saturated fat causes distinct effects on liver and visceral fat accumulation in humans.
“Both groups gained similar weight. SFAs, however, markedly increased liver fat compared with PUFAs and caused a twofold larger increase in VAT than PUFAs. Conversely, PUFAs caused a nearly threefold larger increase in lean tissue than SFAs. Increase in liver fat directly correlated with changes in plasma SFAs and inversely with PUFAs.”
How does that look any good?
Also, SFAs seem to have a pretty bad effect on the gut flora. See for example :
Nutrient signaling: evolutionary origins of the immune-modulating effects of dietary fat
” In a comprehensive literature review, we show that the body preferentially upregulates inflammation in response to saturated FA that promote harmful microbes. In contrast, the host opften reduces inflammation in response to the many unsaturated FA with antimicrobial properties. ”
Again, how’s that any good?
I don’t see any reason why we should tell people to eat more SFAs. Where are those studies that show any improvement from eating more SFAs?
You seem to hang on every possible little details (micronutrient deficiencies, PUFAs, HDL, inflammation) etc, that are all much less well supported that is LDL, and yet you just can’t seem to accept that high-LDL is the primary causative factor, when it’s the most well supported theory so far. There is no other hypothesis out there with as much convincing data and a convergence of all the evidences going its way as strong as this one. There is evidences from about every level of evidences (ecological, biological, experimental, drugs trial). Why is it that you just can’t accept this? What do you gain in having the LDL theory proven wrong? I’m just curious.
Why does the rupture of a plaque always cause a blood clot? What is the clotting mechanism by which platelets are affected by the contents of the plaque?
One of the earliest treatments was warfarin; though superceded as a sole treatment, it must have worked, and no doubt helps to account for part of the drop in CHD mortality during the 1970s, along with reductions in smoking, improvements in workplace and environmental safety standards, and more varied diet due to increased transport and cultivation of non-seasonal or non-regional foods.
George
Many plaque ruptures don’t cause blood clotting. In fact, many plaques appear to heal without any serious consequences. It’s a balance between factors that increase or decrease clotting activity. If the anti-clotting mechanisms are stronger the plaque will just heal and probably go unnoticed.
The anti-platelet activity of aspirin leads to less risk of blood clotting which is probably the main reason why the drug improves prognosis in CAD.
Thank you. I guess the blood clots for the same reason it would with an external rupture like a cut?
The statement “There is strong evidence that lipoproteins play a fundamental role in atherosclerosis and their interaction with the arterial wall appears to initiate the cascade of events that leads to atherosclerosis.” is ambiguous – though I’m sure you didn’t intend it, this could be read as saying that the idea that lipoproteins initiate the cascade is based on strong evidence.
Whereas it seems to me that no-one has good evidence of an initiating process. The idea that failure of eNOS is an early occurrence in endothelial dysfunction preceding plaque formation is consistent with some facts and can be demonstrated in mice; it is hard to see why LDL would play more of a role in this than blood borne toxins, hormones, sugars, and other factors with access to endothelial cells.
eNOS inhibition plays a role in vascular changes in chronic hepatitis, hepatocytes have LDL receptors, but no-one has ever suggested that LDL is a cause of liver disease and indeed all associations run very much the other way.
The likelyhood (it seems to me) is that LDL is a factor that is taken hostage by the process of atherosclerosis, just as red and white blood cells are. More small dense LDL makes the situation worse, and oxLDL may also be a marker of the conditions (oxidising toxins, inflammation, and nutritional deficiencies) likely to initiate endothelial dysfunction.
I guess you’re right George. What initiates atherosclerosis is not entirely clear and there are many factors involved. Maybe it would be more correct to say that lipoproteins are found within the vessel wall early in the atherosclerotic process.
Dr. Greger has stated that indigenous populations in Africa (excepting the Masai) had LDLs between 50-70 and virtually zero incidence of heart disease. He suggests that one can almost be bulletproof, heart-wise, by maintaining ultra-low LDLs while following a plant-based diet that results in similar LDLs. Do you agree? Is there controversy over this, or is this pretty well established?
And on a personal note, how can VLDL-3 be reduced though diet or lifestyle? I’m vegan and don’t drink, but yet I struggle to get below 14 (but other lipids are fine).
Thanks much.
Russell
There is evidence suggesting that exposing someone to low LDL-C through a lifetime reduces CVD risk. For example, nonsense mutations in a specific gene located on chromosome 1 have beens shown to cause low levels of PCSK9. Low PCSK9 will lead to less breakdown of LDL-receptors causing these people to have very low levels of LDL-C. At the same time these individuals have a very low risk of CVD. https://www.docsopinion.com/2012/11/08/pcsk9-a-new-target-for-the-treatment-of-heart-disease/
That’s very helpful thank you.
Also any thoughts on how to reduce VLDL-3?
Age 53, only RF is age/ gender.
I had a CT calcium score 2.5 years ago of 41 putting me at the 80th %tile. Then a CT cor angio with small soft plaque at the proximal LAD about 20%, but I’m RCA dominant.
My LDL was 131 and TG 181 and started statins, since then LDL to 73. Weight down by 30 lbs. Waist from 36 to 32.8 inches.
Now after 6 weeks of LCHF diet (P100/ C 20/ F 210), Labs–>
TC 137–> 204
LDL ( 127 bad
HDL (> 40) 54–> 59 good
TG about same at 87. good
HSCRP ( 0.26 good
Chol/ HDL ( 3.46 good
TG/HDL ( 1.42 good
HDL/TC (> 24%) 39%–> 29% good
Lip(a) ( 3.55 good
A1C 5.6 good
Insulin (<3) 2.5 good
C-pep (0.4-2.1) 0.76
ApoB (<80) 100 Bad
Weights cardio 250 minutes weekly
I'm clearly concerned about the possible worsened risks from these numbers esp apoB and LDL given subclinical CAD (per imaging); although reassured by the rest of the parameters being good (some trending badly)
As per Phinney noting that intial phases of LCHF can raise transiently LDL I will recheck labs in 3 months, but wondered what your thoughts are. I'm hoping this diet won't be unsuitable for me.
I do take ASA 325 3x a week and pravastatin 40 qd.
Thanks…
Low carb moderate fat. Why the high-fat? Eat more fish and soluble fiber. I have high LDL and my spouse has low LDL, eating red meat, etc. while I struggle. You can’t overturn your genetics, I am trying and it’s a challenge.
Axel F. Sigurdsson MD Thanks for a great informative article.
Without getting into professional conflicts, hopefully, I wish to present your article to my GP and Cardiologist who believe that “LDL is simply LDL”.
Without the list of references you refer to in your article, they may disregard your information as unsupported.
Firstly, is it reasonable and acceptable to you, to go ahead with my proposal?
Secondly, would you please publish your list of references?
Respectfully, Ken McMurtrie
Hoping for the reference list to be published to strengthen approach to my GP and Cardiologist with your information.
Would that be appropriate?
In the above bell-shaped curve, notice when Total Cholesterol is less than or equal to 150, the incidence of CAD is very low even for an extended period of time.
This is a very important point to keep in mind and this level is achievable with a healthy diet as my own personal experience indicates.
Axel F. Sigurdsson, MD:
It is a pleasure to review your article on overly simplified Lowering LDL as the target being adopted by (say) 99% physicians or cardiologists in the USA. (European has a different opinion on LDL only approach.)
HDL subtypes (HDL1,2,3,4) or (HDL1,2,3) are related to the statin dose being taken by patients. I have fortunately retained experiences on higher dose on statin with reduced HDL1. HDL1 matures may rely on time and higher does of statin shorten
required growth period of HDL1. While HDL1 + HDL 2 are more effective to clean serum LDL. Aeorobic exercise appears one easy way to increases HDL quantity.
LDL also has subtypes of (LDL1,2,3) while LDL3 being the smallest or more aggressive to permeate into endothelial layer of artery to start “form cells”. Volume of antioxidant in diet appears lower OxLDL. Rush University’s MIND diet appears a better suggestive choice or easier one to adopt. Role of polyphenolic compounds to simulate eNOS to reduce OxLDL penetration has many overlooked benefits. High ROAC food or diet also reduces OxLDL.
Lipid is not the only one response for hardening arteries. The incomplete methylated protein share equal or more vascular lesions. Since higher vitamin (B12, B9, B6 (folate) usually increase protein methylation and lower Homocysteine. In dementia (AD or PD), Tau formation relates to incomplete protein methylation. [ Beyond elevated homocysteine, there are ways to inhibit Tau formation, s.a. CoQ10, Lithium, etc..] None of them has any incentive to pharmaceutical industry or any supportive to widen its acceptance, so are poorly continued educated practice physicians by Pharma’s representatives.
Current widely adopt statin is a knife with three edges: lower LDL, inhibit CoQ10 and cell membrane protective acid sorry, forget the name). Based on AHA statistics, MI patients have a (basic) life expectancy of 17 years. Stain leads a role on congested heart disease in later stage (due to suspended COQ10). Cancer is the second fatality cause in US top 10 causes. Stain also plays a role of faster mutated cells or roots to cancers.
Many large scale elderly studies indicates people who do not take statin with LDL 130+- live longer, while people who take statin with a LDL 110+- live longer.
The misled LDL 70 or lower in AHA ATP III appears only consider that 17 years and not beyond.
It is sad to physicians and patients of misled LDL as the only target.
Thank you very much for your rational and wish more health providers being open their minds to learn beyond than just a box.
R.Y.
mistyped Vitamin (B12, B9 (folate), B6)